Output
NanoPrePro produces two types of output files:
Processed FASTQ with per-read annotations
HTML report summarizing pre-processing results
Processed FASTQ
NanoPrePro appends pre-processing annotations to the FASTQ read IDs. These annotations use standardized flags, summarized below:
flag |
regex |
default |
explanation |
|---|---|---|---|
|
-?d+.d* |
0 |
0: unknown; > 0: sense; < 0: antisense |
|
[0|1] |
0 |
0: non-full-length; 1: full-length |
|
[0|1] |
0 |
0: non-chimeric/fusion; 1: chimeric/fusion |
|
-?d+ |
-1 |
-1: unknown; 0: removed; > 0: 5’ adapter/primer location |
|
-?d+ |
-1 |
-1: unknown; 0: removed; > 0: 3’ adapter/primer location |
|
-?d+ |
0 |
0: unknown; > 0: 5’ poly length; < 0: trimmed 5’ poly |
|
-?d+ |
0 |
0: unknown; > 0: 3’ poly length; < 0: trimmed 3’ poly |
Example
@read_1 strand=0.91 full_length=1 fusion=0 ploc5=0 ploc3=0 poly5=0 poly3=-20
AGAGGCTGGCGGGAACGGGC......TTTCAAAGCCAGGCGGATTC
+
+,),+'$)'%671*%('&$%......((&'(*($%$&%&$-((84*
This example shows that read_1 is:
Sense strand (
strand=0.91)Full-length (
full_length=1)Non-chimeric (
fusion=0)Adapter/primer removed (
ploc5=0 ploc3=0)PolyA trimmed (
poly3=-20)
HTML report
The HTML report provides an overview of pre-processing results, including:
Quality score histograms
Proportion of filtered/passed full-length, truncated, and chimeric reads
Simulated real/random adapter/primer alignment results with interactive cutoff exploration
Choosing Cutoffs from Simulated Real/Random Alignment Results
The report includes an interactive plot of simulated true and random alignments.
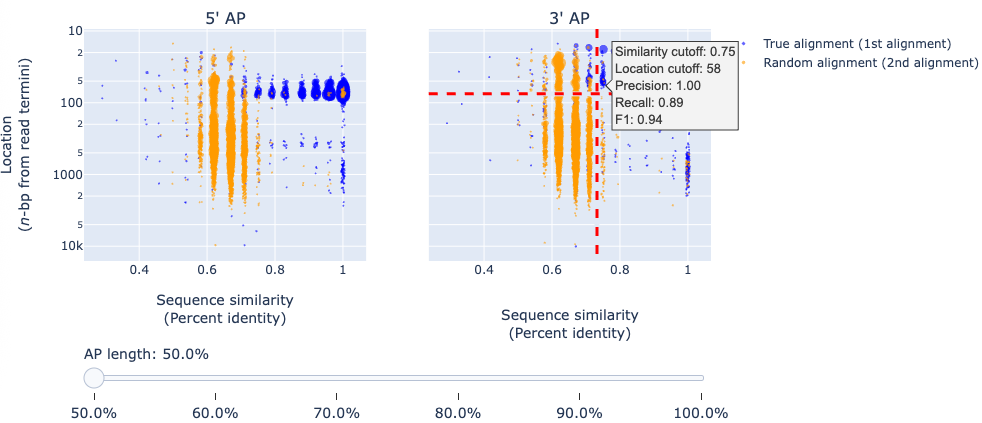
X-axis: sequence similarity
Y-axis: aligned location
Blue dots: true alignments
Orange dots: random alignments
A slider allows switching between results for different adapter/primer substring lengths. Hovering over the plot displays precision, recall, and F-score under specific cutoff settings.
Guidelines
Higher precision: use higher sequence similarity cutoffs and longer adapter/primer substrings.
Higher recall: use lower similarity cutoffs and shorter adapter/primer substrings.
⚠️ Note: Increasing adapter/primer length too far can be counterproductive. Basecalling accuracy often drops near read termini, making the last nucleotides unreliable. Stop increasing the adapter/primer length once most blue dots begin deviating from sequence similarity = 1.